Cluster Chemistry
The reactivity of transition metal cluster complexes has been of special interest in the area of recent organometallic chemistry because of their potential applicability to organic synthesis. The advantageous properties of a multimetallic system over a monometallic one in the substrate activation step are its capability of multiple coordination of the substrate to the metal clusters and the multi-electron transfer between the substrate and the cluster. Transition metal clusters have been, therefore, intensively investigated in the quest to develop new and effective organic transformations achieved by the synergic effect of metal centers. A large number of fine studies on the reaction chemistry of clusters have been reported mainly by so far using metal-carbonyl clusters. However, they sometimes met a serious problem, degradation of the cluster skeleton. This can be utilized for the generation of highly unsaturated metal species from the mother cluster, but it often complicates the reaction, thus impedes the detailed analysis for the role of the neighboring transition metal centers of a cluster.
Polyhydride Cluster
In contrast to polycarbonyl clusters, the trinuclear skeleton of {Cp*Ru(m-H)}3(m3-H)2 (1) is maintained during the reaction even at 180 ˚C. The resistance against the degradation is probably due to the bulky and electron donating nature of the Cp* groups surrounding the trinuclear core. The bridging hydride ligands also play an important role to bind multiple metal nuclei as a tough clamp. In addition, since 1 does not possess an electron-withdrawing group like carbonyl, electron density at a metal center kept in electron-rich. The metal polyhydride clusters are, therefore, expected to be much more active than the metal-carbonyl cluster toward oxidative addition of substrates.
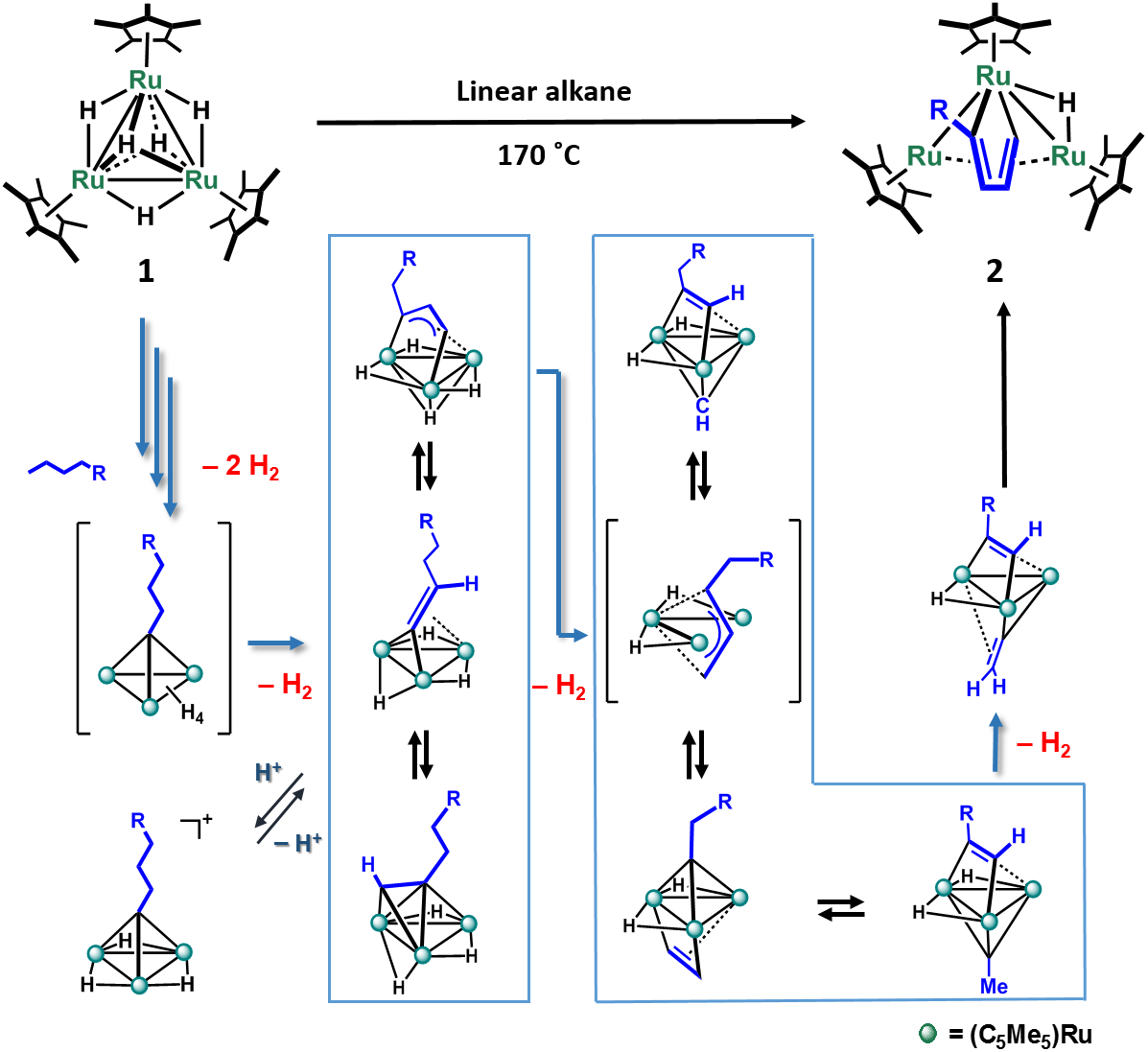
These features enabled us to investigate the reaction of 1 with linear alkanes, which demonstrated that C–H and C–C bonds of alkane were effectively activated on the triruthenium core. We reported the exclusive formation of closo-ruthenacyclopentadiene complex 2 by the reaction of 1 with linear alkane (Scheme 1).1 The molecular structure of 2 represents that totally six C–H bonds on the terminal four carbon atoms were broken during the reaction. However, the mechanism seemed not to be so simple; formation and disappearance of several intermediates were observed during the reaction. Recently, the multi-step skeletal rearrangement of the hydrocarbyl moiety on the triruthenium core has been revealed to proceed as shown in Scheme 1. The reaction scheme is composed of two reversible steps and irreversible steps eliminating dihydrogen. Five molecules of H2 was eliminated from 1 and alkane to form 2. In other words, elimination of dihydrogen from the cluster core seems to drive the transformation of the hydrocarbyl ligands. Such ability to eliminate dihydrogen from the cluster core is crucial property of the polyhydrido cluster composed of Cp*Ru fragments.
by polyhydrido clusters

We applied this ability to dehydrogenative coupling of 4-substituted pyridines (Scheme 2).2 Although addition of oxidants was often required to direct the oxidative C–C coupling, the diruthenium complex, Cp*Ru(m-H)4RuCp* (3), can catalyze the coupling of 4-substituted pyridine without any sacrificial oxidants; the reaction was driven by the spontaneous elimination of dihydrogen from the dinuclear site (though it required above 160 ˚C).
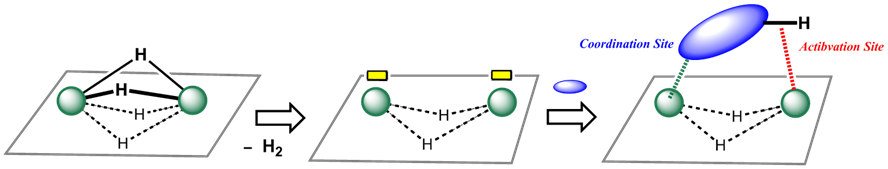
In addition to the dihydrogen elimination, hydride can be also removed from the cluster core via hydrogenation of unsaturated bonds. These processes generate a vacant coordination site on the cluster. In particular, liberation of bridging hydrides provides vacant sites on the adjacent metal centers, and the substrate taken in the reaction site likely interacts with the resulting vacant metal centers all together (Scheme 3). In such a situation, the metal centers probably cooperate in activating the substrate as a binding site and an activation site. This must be the origin of the cluster effect in the substrate activation. We have used the term “multimetallic activation” to refer to a mode of activation achieved as a result of cooperative action of many metal centers.
for Multimetallic Activation
Heterometallic Polyhydride Cluster
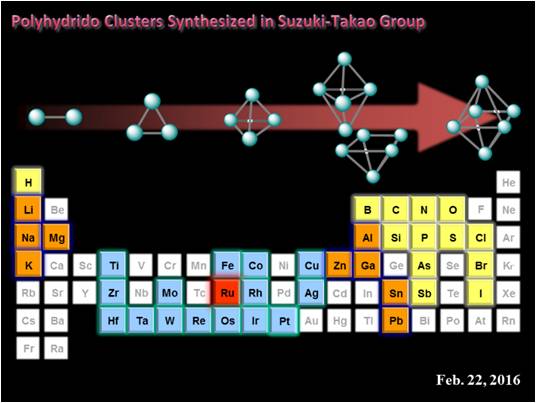
We started the survey of a polyhydrido cluster chemistry using ruthenium in 1988.3 Now, we extends the cluster chemistry to heterometallic polyhydride clusters; the reaction utilizing an anisotropic M–Mʹ bond as well as developments of rational synthetic methods to construct a heterometallic skeleton.4 We succeeded incorporation of various transition metals colored in in the periodic table shown below into the di-, tri-, tetra-, and penta-nuclear skeleton. Enhancement of the anisotropic character of the reaction sites likely causes changes in regio- and chemoselectivity in the stage of substrate activation. Although a number of heterometallic polyhydride clusters have, thus far, been synthesized, there have been only a few reported examples of successful achievement of heterometallic activation of organic substrates.
Cluster-Surface Analogy
As an another important aspect of the cluster chemistry, the relationship of the cluster chemistry with heterogeneous catalysis has been so far taken into consideration. Polynuclear organometallic compounds have been shown to serve as an appropriate model for the adsorbed organic molecules on a metal surface and made important contribution to surface science. The spectroscopic data of the well-defined cluster compounds is often used for the characterization of the chemisorbed species, and the structural data of these compounds provides detailed information about the chemisorbed species. More importantly, information about the reactivity of the chemisorbed species is also provided by the cluster chemistry.
along with redox of the Ru3 cluster

We synthesized a face capping-benzene complex 4, which could be a counterpart of the adsorbed benzene at the 3-fold site of a metal surface (Scheme 4).5 The coordinated benzene provides not only structural and spectrochemical information for the adsorbed benzene, but also important information of its reactivity; we found that the face-capping benzene rotates by 30˚ upon oxidation. The same behavior was also reported for adsorbed benzene molecules on a Rh(111) surface.
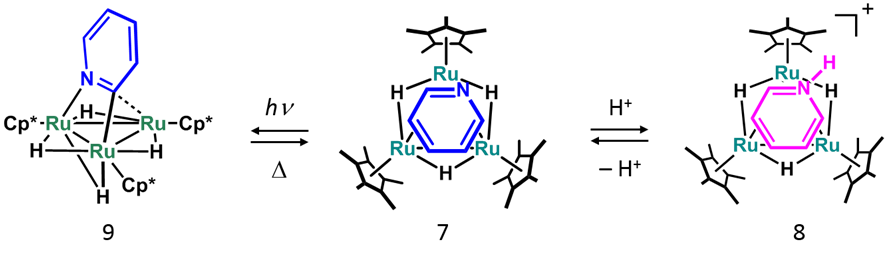
While various adsorption modes of pyridine on a metal surface have been recognized, pyridine has been shown to attach to a multimetallic core in a quite limited mode, mainly through its nitrogen atom. It has been sometimes shown that pyridine is adsorbed on a metal surface with its molecular plane parallel to the surface, but such coordination mode has never been reported in cluster chemistry. We reported the first synthesis of a m3-pyridine complex 7 adopting a face-capping coordination mode (Scheme 5).2b,6 The face-capping pyridine underwent facile protonation at the nitrogen atom leading to the formation of m3-pyridinium complex 8, and the face-capping pyridine ligand was readily transformed into a m3-h2-pyridyl ligand in 9 upon irradiation. These results would be important because of its relation to the hydrodenitrogenation process and poisoning of catalysis.
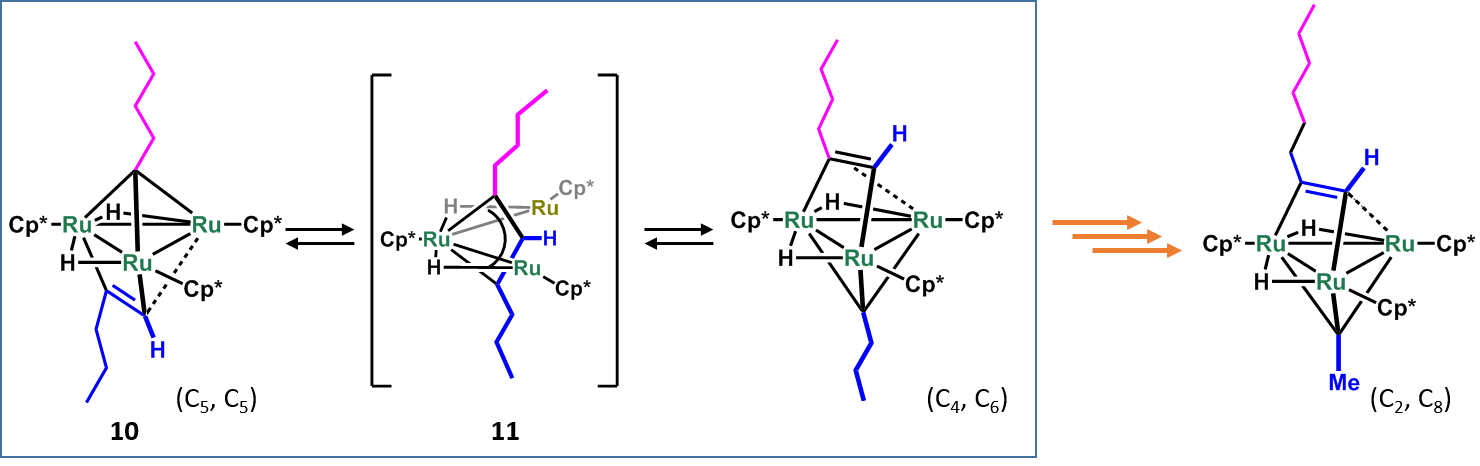
The recombination of a carbon chain, as seen in the cracking of alkane and the Shell Higher Olefin Process (SHOP), is one of the most important processes in the chemical industry. We found that two hydrocarbyl moieties sparated by the Ru3 plane of 10 underwent an unprecedneted interconversion as shwon in Scheme 6, namely two C5 fragments were selectively converted into C2 and C8 fragments on a triruthenium cluster.7 The key-step of this transformation is likely the formation of a m3-metallacyclobutenyl intermediate 11 with cleavage of an M–M bond. These results may also shed light on the mechanism of carbon bond homologation steps on metal surfaces, particularly of those performed on defects, such as steps and kinks.
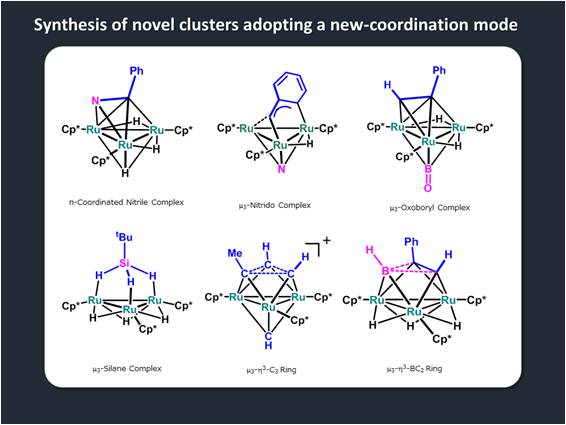
We have also observed several unique and unprecedented structures formed on a Ru3 plane; for example, (⊥)-nitrile,8 m3-nitrido,9 m3-BO,10 and m3-h2:h2:h2-silane complexes,11 and m3-h3-C312 and BC2 rings13 formed on a Ru3 cluster. These complexes are thought to be formed on a metal surface and would play crucial roles in chemical transformations. We continue to research the properties of these exotic species for the purpose of development of novel catalytic reactions using these polynuclear complexes.
References
- a) A. Inagaki, T. Takemori, M. Tanaka, H. Suzuki, Angew. Chem. Int. Ed. 2000, 39, 404–406. b) H. Suzuki, A. Inagaki, K. Matsubara, T. Takemori, Pure Appl. Chem. 2001, 73, 315–318. c) T. Takao, H. Suzuki, Bull. Chem. Soc. Jpn. 2014, 87, 443–458.
- a) T. Kawashima, T. Takao, H. Suzuki, J. Am. Chem. Soc. 2007, 129, 11006–11007. b) T. Takao, K. Kawashima, H. Kanda, R. Okamura, H. Suzuki, Organometallics 2012, 31, 4817–4831.
- H. Suzuki, H. Omori, D. H. Lee, Y. Yoshida, Y. Moro-oka, Organometallics 1988, 7, 2243–2245.
- a) T. Shima, H. Suzuki, Organometallics 2000, 19, 2420–2422. b) T. Shima, J. Ito, H. Suzuki, Organometallics 2001, 20, 10–12. c) M. Ohashi, K. Matsubara, T. Iizuka, H. Suzuki, Angew. Chem., Int. Ed. 2003, 42, 937–940. d) J. Ito, T. Shima, H. Suzuki, Organometallics 2004, 23, 2447–2460. e) T. Shima, H. Suzuki, Organometallics 2005, 24, 1703–1708. f) T. Shima, H. Suzuki, Organometallics 2005, 24, 3939–3945. g) J. Ito, T. Shima, H. Suzuki, Organometallics 2006, 25, 1333–1336. h) T. Shima, T. Ichikawa, H. Suzuki, Organometallics 2007, 27, 6329–6337. i) M. Oishi, T. Kato, M. Nakagwa, H. Suzuki, Organometallics 2008, 27, 6046–6049. j) H. Kameo, Y. Nakajima, H. Suzuki, Angew. Chem. Int. Ed. 2008, 47, 10159–10162. k) T. Shima, Y. Sugimura, H. Suzuki, Organometallics 2009, 28, 871–881. l) M. Oishi, H. Suzuki, Inorg. Chem. 2009, 48, 2349–2351. m) H. Kameo, T. Shima, Y. Nakajima, Organometallics 2009, 28, 2535–2545. n) H. Kameo, Y. Nakajima, K. Namura, H. Suzuki, Organometallics 2011, 30, 6703–6712. o) M. Oishi, M. Kino, M. Saso, M. Oshima, H. Suzuki, Organometallics 2012, 31, 4658–4661. p) M. Nagaoka, T. Takao, H. Suzuki, Organometallics 2012, 31, 6547–6554. q) M. Nagaoka, T. Shima, T. Takao, H. Suzuki, J. Organomet. Chem. 2013, 725, 68–75. r) M. Oishi, M. Oshima, H. Suzuki, Inorg. Chem. 2014, 53, 6634–6654. s) M. Nagaoka, T. Shima, T. Takao, H. Suzuki, Organometallics 2014, 33, 5066–5069.
- a) A. Inagaki, Y. Takaya, T. Takemori, H. Suzuki, M. tanaka, J. Am. Chem. Soc. 1997, 119, 625–626 (1997).b) T. Takao, H. Suzuki, Coord. Chem. Rev. 2012, 256, 695–708.
- T. Kawashima, T. Takao, H. Suzuki, Angew. Chem., Int. Ed. 2006, 45, 7615–7618.
- A. Tahara, M. Kajigaya, M. Moriya, T. Takao, H. Suzuki, Angew. Chem. Int. Ed. 2010, 49, 5898–5901.
- T. Takao, T. Kawashima, K. Matsubara, H. Suzuki, Organometallics 2005, 24, 3371–3374.
- T. Kawashima, T. Takao, H. Suzuki, Angew. Chem. Int. Ed. 2006, 45, 7615–7618.
- T. Kaneko, T. Takao, H. Suzuki, Angew. Chem. Int. Ed. 2013, 52, 11884–11887.
- M. Nagaoka, H. Tsuruda, M. Amako, H. Suzuki, T. Takao, Angew. Chem. Int. Ed. 2015, 54, 14871–14874.
- a) T. Takao, A. Inagaki, E. Murotani, T. Imamura, H. Suzuki, Organometallics 2003, 22, 1361–1363. b) T. Takao, M. Moriya, H. Suzuki, Organometallics 2007, 26, 1349–1360.
- T. Kaneko, H. Suwa, T. Takao, H. Suzuki, Organometallics 2013, 32, 737–740.